“The good cholesterol or Milano A-1 mutation”

The Good: This mutation reduces the risk of arteriosclerosis by increasing good cholesterol levels. Thus, providing carriers up to an 88% lower risk of developing arteriosclerosis and other adverse effects due to high cholesterol, such as the incidence of heart attack and stroke. This mutation has been identified as to have originated and is prevalent in Italy. -1
The reality: While the mutation does provide a benefit of reducing “bad” HDL cholesterol to some trait carriers, the mutation resulted in damage to the gene that controls
(among likely many other things yet to be understood) the regulation of enzymes secreted in the intestines and the liver. How the mutation exactly affects HDL levels is yet not fully understood.-2 The mutation is directly associated with increased renal (kidney) dysfunction. Additional negative clinical manifestations have been identified within the liver, heart, skin, and larynx. The mutation has been associated with neuropathic disorders.-2 Sequencing the coding region revealed a C → T substitution…(which) resulted in a premature (damaged) stop codon-3. “The combination of (a) marked familial decrease of HDL-cholesterol and apo-A-I, (b) hypertriglyceridemia {an abnormal concentration of triglycerides in the blood} (that is resistant to diet and drug treatments), (c) {plus} changes of the HDL composition and apoprotein pattern, (d) normal LPL and LCAT activities, and (e) the absence both of a significant increase of atherosclerotic vascular disease in the family and of cholesterol accumulation in tissues strongly suggests that this is a new disease entity in the field of lipoprotein pathology.”-4. At least 700 inherited metabolic disorders are associated with genetic loss specific to liver function related to this mutated gene.-5
Significant hypertriglyceridemia with a very marked decrease of high density lipoproteins (HDL)-cholesterol levels (7-14 mg/dl) was detected in three members (father, son, and daughter) of an Italian family. The three affected individuals did not show any clinical signs of atherosclerosis (which is “good”, however)…the subjects showed a significant enlargement of the lipoprotein particles and a concomitant increase in the triglyceride content…The observation in otherwise clinically healthy subjects of hypertriglyceridemia, reduced HDL-cholesterol, and marked apoprotein abnormalities, without a significant incidence of atherosclerotic disease in the family suggests this is a new disease entity in the field of lipoprotein pathology, very probably related to an altered amino acid composition of the apo-A-I protein-4
(see Weisgraber et al. 1980. J. Clin. Invest. 66: 901-907).
Mutations in the APOA1 gene have also been linked to familial hypoalphalipoproteinemia (low levels of high-density lipoprotein cholesterol (HDL-C)). Patients carrying (a single) APOA1 mutation typically demonstrate reduced levels of high-density lipoprotein (HDL) cholesterol …(but) the presence of (two) APOA1 mutations generally results in complete absence of HDL cholesterol and may include additional clinical features such as xanthomas (a skin condition in which certain fats build up under the surface of the skin) or corneal opacities (scaring of the cornea).
Mayo Clinic Labs-2
The hereditary…late-onset diseases that show variable penetrance due to amyloidosis (due to the protein misfolding caused by the APOA1 mutation)…present as non-neuropathic systemic amyloidosis (organ dysfunction), with renal (kidney) dysfunction being the most prevalent manifestation.
Mayo Clinic Labs-2
Defects in this (APOA1) gene are associated with HDL deficiencies, including Tangier disease…
https://www.ncbi.nlm.nih.gov/gene/335
Summary: While the genetic mutation to APOA1 does render the benefit of lowered HDL cholesterol which also reduces the risks associated with arteriosclerosis, there are apparent degradative side effects directly related to this mutation. There was not any novel (new) de novo genetic material created. It was a “benefit” derived directly from degradation. While high HDL cholesterol levels are “bad,” having very little (or no) HDL is as bad or perhaps worse. The combinations of the factors associated with the study of this mutation have rendered researchers to conclude that this mutation effectively “forms a new disease entity in the field of lipoprotein pathology)-2. This is not surprising to genetics in the least because mutations cause countless genetic disorders, illnesses, cancers, disabilities like Autism, or deformities. Mutations are much more associated with pathologies than with any supposed beneficial causality. The function of biological systems does not exist “in a bubble” as a single isolated trait, but they cascade throughout many systems that impact-related and unrelated processes.
1- Clinical: Apolipoprotein A-I (APOA1) Gene, Full Gene Analysis, Varies
2-https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2694339/
3- https://www.science.org/doi/10.1126/science.323.5914.583b
5- Pampols T. Inherited metabolic rare disease. Adv. Exp. Med. Biol. 2010;686:397–431. [PubMed] [Google Scholar]
“The unbreakable bones mutation”

The Good: This mutation provides stronger than average bones (higher bone densities) and is therefore dubbed as the “stronger bones” or exaggerated as the “unbreakable bones” mutation.
The reality: Mutations within the gene are known to lead to various bone disorders, including high-bone-mass (HBM), homozygous loss-of-function mutations caused by osteoporosis syndrome (OPPG). This syndrome is characterized by early-onset osteoporosis and complications in eye development. In experiments with mice, the LRP5 mutation expressed a low bone mass phenotype. They also exhibited decreased osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization. Furthermore, associations have also been reported between the LRP5 gene and polymorphisms (negatively affecting bone mass and size) in those affected by the mutation.-1 These mutations can directly cause a loss of function (in the) low-density lipoprotein receptor…(LRP5) of its inhibitor Sclerostin (SOST). LRP5 mutations reduce “Wnt signaling” that causes many disorders, including childhood-onset osteoporosis called “Idiopathic juvenile osteoporosis” (IJO)-2. The first symptoms of IJO appear well before puberty, and the main symptoms include reduced bone mineral density (BMD), vertebral compression fractures, and metaphyseal fractures in the long bones. The fractures lead to bone pain and impaired mobility [1–3]. Researchers report that this mutation exhibits “dangerous and unwanted side effects.” The mutation can be inherited in an autosomal dominant manner (meaning the carriers receive two mutations) which increases the likelihood of degradative side effects-3
Our results provide additional information on the role of LRP5 mutations and their effects on the development of juvenile-onset primary osteoporosis, and hence the pathogenesis of the disorder. The mutations causing primary osteoporosis…may therefore result in decreased bone formation.
All LRP5 mutations associated with primary osteoporosis …(were) located in the coding regions of the LRP5 gene.
Korvala, J., Jüppner, H., Mäkitie, O. et al. Mutations in LRP5 cause primary osteoporosis without features of OI by reducing Wnt signaling activity. BMC Med Genet 13, 26 (2012). https://doi.org/10.1186/1471-2350-13-26
Summary: While the genetic mutation to the LRP5 gene does render the benefit of an increased density in bones which reduces the risks associated with osteoporosis, there are apparent degradative side effects directly related to juvenile-onset osteoporosis as decreased bone development. Additionally, other mutations within this gene affected serotonin synthesis, increased bone fractures, bone pain, and impaired mobility. The function of biological systems does not exist “in a bubble” as a single isolated trait, but they cascade throughout many systems that impact-related and unrelated processes. While higher density bones have some potential benefits, side effects have rendered disorders and juvenile-onset diseases-1.
2-https://bmcmedgenet.biomedcentral.com/articles/10.1186/1471-2350-13-26
3- https://www.science.org/doi/10.1126/scitranslmed.aau7137
“The super-athlete mutation”

The Good: Increased fast-twitch muscle fibers provide quicker movements in elite athletes. Carriers of this mutation potentially have exceptionally well-developed fast-twitch muscles, leading to faster and stronger athletes. The mutation was discovered more frequently among elite athletes.
The reality: The ACTN3 mutation is directly associated with the absence of α-actinin-3, which leads to loss of muscle power and strength, is determined to be toxic, detrimental to force generation, and performance-enhancing isoforms (harmful to muscle function), reduced bone and muscle mass increasing susceptibility to injury. Linked to an early onset of Pompe disease, decreased muscle strength in boys with Duchenne muscular dystrophy, and a slower, more oxidative muscle profile in people carrying two copies (of the mutation). The mutation has been linked to increased muscle glycogen, usually a rare condition that causes a Glycogen Storage Disease-2. Other adverse effects include hypertension, hyperkalemia, acidosis, and expression of structural and oxidative signaling proteins associated with functional or sensory loss in neurodegenerative diseases-3. Also, an imbalanced metabolism and excess reactive oxygen species (ROS) generation end in a range of disorders such as Alzheimer’s disease, Parkinson’s disease, premature aging, and many other neural diseases are associated with this mutation, and more.-4. There has also been an association between the mutation and acute mountain sickness due to a decreased rate of acclimatization, making the mutation carriers more susceptible to the effects of hypoxia during the acclimatization process and may develop AMS symptoms due to “decreased pressure of oxygen in the arterial blood.” These result in adaptation compensations that emerge as decreased oxygen raised heart rates and cardiac output, headache, fatigue, dizziness, anorexia, and nausea.-5
“The ACTN3 gene encodes for α-actinin-3, an actin-binding protein that is specifically expressed in fast skeletal muscle fibers.” Excessive α-actinin-3 expression such as seen in this mutation have “a “toxic” effect on skeletal muscles.”
“Contrary to expectation, in vivo “doping” of ACTN3 at low to moderate doses demonstrated an absence of any change in function. At high doses, ACTN3 is toxic and detrimental to force generation…detrimental for muscle function.”
Gene transfer overexpression due to the ACTN3 mutation…”did not enhance muscle mass but highlighted the primary role of α-actinin-3 in modulating muscle metabolism with altered fatiguability.”
“Our findings demonstrate the sensitive balance of sarcomeric α-actinin expression…(the) overexpression of ACTN3 reveals insight into its metabolic role in skeletal muscle, but this is innately linked to the level of expression which can be easily disrupted causing detrimental functional effects, reminiscent of disease.”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5986729/
Summary: While the genetic mutation to the ACTN3 gene does at least anecdotally render the proclaimed benefit of an increased force generation in muscles, proof for this claim has not been identified if they exist. The ACTN3 mutation has been determined to impact the a-actinin-3 that modulates muscle metabolism directly. It appears the specific benefits claimed by this mutation are largely yet unidentified, but the clear tendencies for side effects are clear and measurable. Higher levels of a-actinin “may increase muscle mass and force-producing capacity (in one) deficient in such skeletal muscle…causing detrimental functional effects, reminiscent of disease.”-4. Contrary to expectations, doping increased levels of a-actinin are detrimental to force generation and overall muscle function.
1-The Effect of ACTN3 Gene Doping on Skeletal Muscle Performance;
2-https://www.hopkinsmedicine.org/health/conditions-and-diseases/glycogen-storage-
4- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2724665/
5- https://genesenvironment.biomedcentral.com/articles/10.1186/s41021-019-0133-8
“The Sickle Cell Mutation or Malaria Resistance”
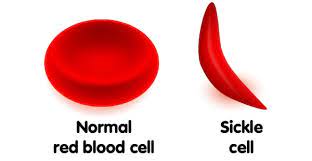
The Good: The Sickle Cell protein mutation leads to an abnormally shaped red blood cell to emerge not round but in a deformed sickle shape. The Sickle Cell mutation affects the beta-globin (HBB) gene and has also been found in at least two other pathways, including encoding hemoglobin S and C in the beta-globin. This mutation directly allows those affected to have a 29% – 93% survival rate of Malaria-1 because it did not recognize the mutated red blood cell proteins as food. Therefore, counter-intuitively, the misfolded sickle-shaped red blood cells provide a survival benefit that permitted millions of human beings from death by the horrible disease of Malaria.
The reality: Sickle Cell is an inherited blood disorder or disease brought about by a genetic mutation that allowed carriers to survive Malaria. The mutation is known to cause severe compilations in those that carry the trait. The disease can be anywhere from painful to deadly. Other side effects include Acute Chest Syndrome, stroke, learning disabilities, heart problems, kidney problems, and other degradative conditions including anemia, pain, swelling of hands and feet, increased rates of infection, delayed puberty or growth, pulmonary hypertension, multiple organ damage, loss of body water in urine, blindness, leg ulcers, gallstones, priapism (obstruction of blood vessels in the penis), and other vision problems. Symptoms include fever, pale skin, yellowing (jaundice) eyes, stroke, trouble walking, confusion, and more. Sickle Cell also increases blood pressure and blood clots, especially during pregnancy increasing miscarriage, premature births, and low birth weights.-3 The mutation persists today as an endemic disease which means it cannot be removed from the genome and will persist in humankind, never to be “corrected” (removed).-2 Also, the mutation is associated with splenetic sequestration (pooling), and other disabilities and conditions.-3
As with all inherited genetic diseases, you’d expect natural selection to week out a gene that has such unpleasant consequences but with sickle cell disease, that doesn’t seem to be the case…Indeed, as of 2015, about 4.4 million people have sickle cell disease, while an additional 43 million have sickle cell trait.-3
…it is simply amazing to me that this all comes from one very small change, changes in (a single) amino acid…has big effects on all of the other systems in the body that the red cells pass through.-4
Sickle cell disease is a hereditary…caused by mutations in one of the genes that encode the hemoglobin protein…The mutation causes the red blood cells to take on an unusual sickle shape. Individuals affected by sickle cell disease are chronically anemic and experience significant damage to their heart, lungs, and kidneys.”-4
Summary: Clearly, not dying of Malaria is a benefit! Perhaps many millions have survived this horrible disease due to this mutation. However, the mutation caused damage to the red blood cells, which inadvertently provided this benefit. Sounds like an excellent example for universal common descent evolution (UCD)? Well, it is not. Let’s first recognize (as noted in the quote above) that natural selection cannot, contrary to expectations, remove “bad” genetic consequences. This is important to keep in mind when concerning virtually every known mutation directly associated with disease or death. Supposed “good” mutations are exceedingly rare–if they exist at all! If UCD is happening, it must have used an unknown mechanism to clean up all the bad genetic mistakes. However, like those supposed for UCD, any such mechanism remains at large and never observed. These negative genetic consequences due to copy error mutations are called endemic. Endemic means they cannot be reversed in the gene pool and is pervasive in regions or specific populations. Sickle cell was caused by a mutation in the gene that folds for red blood cells that also became part of gametes in the reproduction system of humans. This mutation typically caused round-shaped red blood cells to emerge as deformed sickle-shapes. Because round-shaped red blood cells were the food source for the parasite that causes Malaria, the deformed red blood cells were now effectively invisible to the parasite. Therefore, this mutation caused rigid, sticky, and misshapen (deformed) red blood cells, which counter-intuitively prevented Malaria and saved many lives. However, calling this a “beneficial” mutation does require a strong prescription for rose-colored glasses. This mutation is perhaps the best example of copy errors rendering benefit. However, this was a massive genetic cost of disease, suffering, illness, and death. Like all the other supposed beneficial mutations, the resume is quite ugly, but sometimes even broken things can benefit: Even a broken clock is correct twice every day.
1-https://bigthink.com/surprising-science/evolution-is-still-happening-beneficial-mutations-in-humans/
2- Complications & Damage – Sickle Cell Speaks ;
3- https://www.mayoclinic.org/diseases-conditions/sickle-cell-anemia/symptoms-causes/syc-20355876
4-https://www.genome.gov/genetics-glossary/Sickle-Cell-Disease
“The super oxygen saturation mutation”

The Good: Increased oxygen saturation rates at higher altitudes. The native population of the Andean Highlands of Quechua, Peru, and other groups is thought to have increased exercise capacity due to this mutation. This mutation has allowed those that carry the trait to exercise in lower oxygen and high-altitude environments. The mutation modifications to the gene are thought to enable people to thrive at high altitudes without experiencing altitude sickness.
The reality: The idea of this beneficial mutation is enticing but “…direct evidence for genetic adaptation is lacking, and only limited data exist linking genotype to phenotype. One of the largest studies involved (only) 26 Tibetans-1. EGLN1 is found in cell ovarian cancer and melanoma when combined with higher HIF1A levels, a known side effect of this mutation. Decreased oxygen concentrations lead to increased glucose uptake.-2 Finally, the study found that only 0.25% (1/4 of 1%) indicated the proclaimed mutative benefits were essentially non-existent in the sample. Leaving researchers to write: “Quechua provide support for the hypothesis that Andeans are genetically adapted to altitude…was not definitive on the issue of genetic association“. -3. Also, six other cancers associated with a mutation to this gene include breast, kidney, cervical, paraganglioma, adrenocortical, and Waldenstrom’s Macroglobulinemia.-4.
…we demonstrated that the EGLN1 haplotype in Tibetans is associated with (improved oxygen saturations)… we hypothesized that this haplotype might be associated with an EGLN1 gain-of-function mutation …(h)owever, our initial functional assessment…associated (the mutation) with augmented rather than decreased erythropoiesis…caused by a missense mutation…“-1
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4473257/
To illustrate the cascade of areas the EGLN1, here are some of the many biological functions in which it is used: “cardiac muscle tissue morphogenesis, cellular response to hypoxia, cellular response to stress, cytoplasm, cytosol, heart trabecula formation, iron ion binding, L-ascorbic acid-binding, labyrinthine layer development, negative regulation of sequence-specific DNA binding transcription factor activity, nucleus oxidoreductase activity, acting on single donors with incorporation of molecular oxygen, incorporation of two atoms of oxygen, oxygen homeostasis, peptidyl-proline 4-dioxygenase activity, peptidyl-proline dioxygenase activity, peptidyl-proline hydroxylation to 4-hydroxy-L-proline, protein binding, regulation of angiogenesis, regulation of transcription from RNA polymerase II promoter in response to hypoxia, response to hypoxia, response to nitric oxide, ventricular septum morphogenesis, zinc ion binding.”-4
Summary: This proclaimed “beneficial mutation” seems to have some laboratory plausibility as the positions on the gene are associated with known cases of hypoxia. However, in even the most prominent groups, the populations studied were relatively small and diverse, consisting of only 26 individuals. Of those studied, it was estimated that only the tiny percentage carried any such enhanced physical characteristic. The research left scientists concluding that those with this mutation thought to be “genetically adapted to higher altitude…was not definitive on the issue (based on) genetic association.”-1. The emergence of this vastly rare mutation was determined to have come from a “missense mutation” (a mistake in the DNA that results in the wrong amino acid being incorporated into a protein), causing deformation and degradation of the protein. Ultimately, this supposed beneficial mutation failed to provide adequate evidence of any new genetic information or trait. It offered evidence of disfunction and potentially vastly rare (really anecdotal) protections from polycythemia (an increase in the number of red blood cells in the body associated with high altitude sickness or hypoxia). The mutation has also been associated with various cancers, including “Breast, Kidney, Cervical, Paraganglioma, Adrenocortical, and Waldenstrom’s Macroglobulinemia.)-4
1-https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4473257/
2-https://cancerres.aacrjournals.org/content/79/10/2564.short;
3-https://www.pnas.org/content/116/48/24006;
4-http://www.cancerindex.org/geneweb/EGLN1.htm
“The divers (breath-holding) mutation”

The Good: Increased time of holding breath allowed those with the mutation to become mutationally more adept at diving by staying underwater longer. The modification might enable carriers to hold their breath for very long periods compared to the average. This mutation has been found most common among the free-diving Bajau people.-1
The Reality: The actual rate of this mutation is elusive at best and any supposed benefits are anecdotal and very rare. The mutation is most commonly associated with hearing loss.-2 This mutation is associated with both pre & post-synaptic molecular changes in the brain called substantia nigra damage-3 which are directly associated with memory-6. Damage caused to the dopaminergic neurons within the midbrain is associated with Parkinson’s Disease, movement disorders, Huntington’s Disease, tremors, and rigidity. Also, white and gray matter changes within the striate cortical pathways.”-2 Diseases associated with PDE10A include Dyskinesia (involuntary, erratic, twisting movements of the face, arms, legs, or trunk), Limb And Orofacial, Infantile-Onset, and Striatal Degeneration (a neurologic disorder characterized by delayed motor development), as an Autosomal Dominant disorder (pattern of the inheritance of some genetic diseases such as Marfan’s Syndrome)-4. Ultimately this mutation negatively affects the “…region of the brain that contributes to controlling movement and cognition.” Degradations due to the CX26 mutation include KIDS Syndrome and deafness. Also, it causes sleep apnea, breathing problems, and sensitivity to Carbon Dioxide.-5.
PDE10A is (an) “autosomal dominant disorders, (and) only one allele of a mutated gene is necessary for disease.”
Mechanisms and Morphology of Cellular Injury, Adaptation, and Death; Margaret A. Miller, James F. Zachary, in Pathologic Basis of Veterinary Disease (Sixth Edition), 2017
We found that PDE10A and ADCY5 mutations were associated with decreased PDE10A expression in the striatum and globus pallidus (brain matter), decreased dopamine transporter expression…loss of substantia nigra neuromelanin-containing neurons, and microstructural white and gray matter changes…”-2
Summary: The mutated sequence associated with increased oxygen saturation rates has been identified and lab-verified, however, this mutation’s proclaimed benefits are rare (if they exist at all). Conclusions are anecdotal as finding carriers also with the proposed benefit of holding their breath longer have been ellusive. Carriers and their offspring are often plagued with many dangerous side effects, including harmful effects to the region of the brain that controls movement, cognition, and memory. It can cause many adverse effects when the trait is passed, including diseases, deafness, and neurological disorders. Conditions include Huntington’s, KIDS Syndrome, Infantile-Onset, and Striatal Degeneration. Ironically, the mutation might directly associate breathing problems, including sleep apnea and increased sensitivity to carbon dioxide.
2 https://movementdisorders.onlinelibrary.wiley.com/doi/abs/10.1002/mds.27523
3 https://www.sciencedirect.com/topics/medicine-and-dentistry/substantia-nigra
4- https://www.sciencedirect.com/topics/medicine-and-dentistry/autosomal-dominant-disorder
5- https://www.newswise.com/articles/genetic-mutation-may-hold-answers-to-controlled-breathing;
6- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6632334/
“The less sleep or Edison mutation”

The Good: The gene thought to regulate sleep duration has the proclaimed benefit of requiring fewer hours of sleep for those with this genetic mutation. Popularizers of this beneficial mutation claim that sleeping for 7 to 8 hours per night (such as is the average) is a disadvantage compared to those that require fewer hours of sleep. The mutation allows (or more accurately causes) its carriers to function “normally” on only 4-5 hours of sleep per night. Some say perhaps Thomas Edison had this same mutation.-1
The reality: It is subjective to conclude that less sleep is better or regular. This mutation is known to directly cause the deletion of orexin neurons (receptor genes), resulting in narcolepsy. Also, it causes orexin signaling that negatively affects maintaining arousal (wakefulness) and sleep consolidation (quality sleep). Furthermore, the administration of orexin neurons experimentally increased arousal and decreased sleep duration. “Therefore, our findings provide insight into how orexin expression, which is involved in many biological functions, is regulated at a molecular level. We previously reported that the human DEC2 mutation contributes to short sleep duration in humans. Research also found decreased sleep durations in other animals, including flies and mice.-1. Regardless, “…the detailed mechanism of this sleep phenotype has remained elusive.” Clock genes, such as this mutation, are involved with tumor progression, playing an “important role in circadian rhythm, cell proliferation, apoptosis, hypoxia response, various stresses, and epithelial-to-mesenchymal transition (EMT) of tumor cells” -2. Therefore a mutation likely will manifest negatively within these areas as well. This negative feedback system plays an essential role in 24-hour rhythmic regulation for circadian rhythm. The disturbance of circadian rhythm may induce metabolic syndrome, diabetes, Alzheimer’s disease, depression, sleep disorder, or cancer. Less sleep or nighttime work may increase cancer risk due to melatonin suppression. Mutations in the DEC2 gene are involved in the human short sleep phenotype and the regularization of sleep duration in mammals. Fewer hours of sleep are directly associated with excessive daytime sleeplessness, especially in children. Excessive sleepiness is associated with a 60% higher incidence of cancer and an 80% increased risk to children with neoplasm in the central nervous system. Sleep disorders account for 40% of the entire children with cancer! Also, the mutation is associated with the expression of DEC1, which is associated with human oral cancer cells, breast cancer cells, and pancreatic cancer cells.-2
All living beings from bacteria to human have circadian rhythm, which is dominantly regulated by clock genes (DEC2). The molecular mechanism of circadian rhythm depends on negative feed-back system by clock genes.”-2
(Mutated) Clock genes, major regulators of circadian rhythm, are involved in tumor progression…cell proliferation, apoptosis, hypoxia response, various stresses, and…tumor cells.“-2
Summary: The premise that less sleep is a benefit is very subjective. The quality of sleep and resulting wakefulness are more objectively positive attributes of sleep. First, any associated advantage from this mutation is not clear to researchers, concluding that any good benefits behind this proclaimed good mutation “remain elusive.” Perhaps the most critical role of quality sleep is found by maintaining good circadian rhythms. Good circadian rhythms are directly associated with good health and reduced disease. Also, the mutation is associated with many vastly harmful side effects, including narcolepsy, poor quality of sleep, diabetes, Alzheimer’s Disease, depression, sleep disorders, and even increased risk of developing cancers including breast and pancreatic. Children with this mutation have an 80% increased risk of developing central nervous system cancer, while any sleeping disorder in children increases cancer risks by 40%.
1- DEC2 modulates orexin expression and regulates sleep
2- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4716847/
“The Ivory Tusk loss Mutation in elephants”

The Good: It is estimated that 35,000 elephants are killed each year for their ivory, so a reduction helps protect these threatened populations.-1 The female elephant, as measured since the 1940s, has a higher likelihood of being born “tusk-less” from about 18.5% to about 33% today. Tusks are associated with the teeth genes and are essentially overgrown molars used for a wide variety of tasks. Since poaching has increased, it is believed that this mutation has caused this result to benefit the elephant population at large as a form of natural selection.
The reality is tusks are genetically passed by heredity. Any male born without tusks is clearly a degradative disability that leads to death. Due to changing environments, tusks are not needed by females. Also, poaching impacts the genetic pool at reproduction. Those carriers of the tusk gene are more likely to be killed than the tuskless. “They have this very compelling genomic data…(but) Scientists are still not sure which changes are causing a loss of tusks in either of the genes… “It appears that natural selection (as driven by gametes at reproduction) is the best and only answer as to why there has been a measurable reduction in the number of female elephants born tusk-less. The mutation has been isolated as a degradative or disability in male elephants. There leaves little reason to believe that it is also a handicap for females—just not (usually) deadly. The findings here are largely anecdotal as the prevalence of tusk-less females has always been measurable. The rapid reduction of elephant populations combined with an ever-changing environment has led to a “bottleneck that strongly favors (the) tusk-less phenotype.”-1. Think about it this way: the elephants (primarily male but also females) with tusks are poached and killed. Those females that carry the tusk-less phenotype live and reproduce. Thus expanding this recessive trait with each passing year by genetic bottlenecking. This is natural selection (by environmental pressure) as a direct result caused by reckless human beings. The results are not due to mutations but are heredity favoring the recessive trait of females that are tusk-less. Finally, while there are anecdotal reports of tusk-less males in the populations of elephants worldwide, these are plausibly explained by either rare exceptions such as injuries or perhaps observer error–not beneficial mutations.
Researchers at Gorongosa National Park “noticed that the elephants with no incisors were usually female. The park has never seen a tuskless male, suggesting the trait related to tusklessness is sex-linked.-2
If a female elephant had one copy of the tuskless mutation, they would have no tusks. So, when the elephant reproduces, half of their daughters will have tusks, and the others will not have tusks at all. Half of the males will have tusks if their offspring is male, and the other half will die, possibly even before birth…”-2
Summary: Tusks are detrimental to male elephants used for various purposes but certainly as the primary form self-defense that includes the ability to gain a mate and reproduce. Tuskless male elephants face certain death. This supposed beneficial mutation has helped female elephants escape ivory hunters, and natural selection, not conversions, has likely played a role in this reality. Elephant parents carry hereditary traits (alleles) to pass tusks and tusklessness to offspring. Over recent decades, studies have found that due to various factors, including changing environments and ivory hunting, allele frequencies of female tusklessness have increased from 18.5% to 30%. This is evidence reflects natural selection pressures that are likely impacted by ivory hunting. Therefore, the benefit claims seem validated, but it is not. Why? Because identifying the genetic sequence of the gene responsible for female trustlessness is not due to any copy error mutation but are gene variants provided by the parents that are passed at reproduction. Because many tusked females have been hunted and removed from the gene pool, tusklessness has increased. It is that simple. Like brown or blue eyes in human beings, these are gene variants–presuming them to be due to an ancient mutation is not verifiable and is ultimately speculation. Any assumption of this occurring by a supposed beneficial mutation is based on ancient premise because we have always observed both tusk and tuskless female evidence.
1- https://africa.si.edu/collection/conservation/protect-ivory/
“Less Wisdom Teeth Mutation”
The Good: The presumption is that the jawline has become smaller (due to another mutation, MYH16), which is said to have restricted the human jaw size, also related to supposed growing brain size. Some have evolved to get fewer wisdom teeth as an act of natural selection. The belief reasons that as we continue to grow, humankind will not have any wisdom teeth one day. Today the presumption is this random gene mutation occurred nearly 400,000 years ago or longer. Those born with this mutation in the gene for teeth have gained a beneficial attribute of requiring fewer wisdom teeth extractions.
The reality: This genetic trait is hereditary and is far from being unusual. This gene affects about 20%-40% of the entire human population who get less than four wisdom teeth. Some humans get double wisdom teeth—up to eight teeth.-2 The most obvious answer is natural selection and the operation of heredity. Perhaps over the centuries, isolated gene sequences that include teeth have been turned off. It mi ht be as simple as we do not need wisdom teeth as we did hundreds of years ago. However, at best, misinformation is associated with a spontaneous mutation of the distant past. The reality is geneticists can identify where the genetic sequence resides that accounts for wisdom teeth but there is no ancient DNA sample to compare against to confirm this speculation. The mutation that is supposed to reduce the number of molars (wisdom teeth) is are deduction as to what might have happened in the distant past. This claim is not empirically based but are speculations on the genetic evidence and not from genetic evidence. “Are wisdom teeth genetic? The answer is complicated. Ances ryDNA looks at…genetic markers related to wisdom teeth to estimate whether or not you developed all four wisdom teeth. (Howe er), there are still a lot of unknowns about the role genetics plays in tooth development — particularly wisdom teeth because they aren’t present at birth like the rest of your teeth are. As genetics are complicated, and there may be other factors at work besides just your genes, this trait is hard to pred, So even if your genetic markers predict an absence of wisdom teeth, you may still have them.”-3. In other words, claiming a beneficial mutation as the cause behind reduced wisdom teeth production is highly dubious, not explained by evidence but, as it turns out, a math formula.
Perhaps the greatest significance of the study is that it demonstrates how the evolutionary development of our bodies can be described mathematically…Having a formula to describe these features could help us understand some of the greatest mysteries of our evolutionary history.”-2
Summary: The gene sequences that control wisdom teeth are genetically passed as variants derived from the parents. As we have pointed out repeatedly, identifying the genetic sequence does not provide evidence for an ancient mutation for elements of heredity. Heredity emerges from the sex cells of the parents that provide variants the ultimately form gametes that create the offspring at conception. This process has nothing to do with copy error mutations. Any presumption of an ancient mutation cannot be verified because no such DNA sample can verify a pre-mutated sample, rendering this as speculation. It is possible that due to changing needs for molars, slight changes have been made due to natural selection. Any assumption of this occurring due to a beneficial mutation is based on speculation because, as far as we know, humans have always had various genes that affect wisdom teeth production.
1-https://en.wikipedia.org/wiki/Wisdom_tooth;
3-https://www.ancestry.com/lp/traits/wisdom-teeth
“Blue Eyes Mutation”
The Good: The presumption is that the emergence of blue eyes in human beings happened by the mutation in a single human ancestor about 6,000-10,000 years ago. There ore, those born with this mutation in the gene for eyes have gained the attribute of having blue eyes which is considered as being neither a positive nor negative trait.-1
The reality: Hereditary function finds that the sex cells of the parents produce gametes that emerge as various traits in the offspring called allele frequencies. Some of these expressions (traits) are dominant because they happen more often, while others are recessive because they emerge less often. Some aspects of eye color function by a pigment called melanin that produces the brown color. Lower melanin has less pigment and, therefore, green, hazel, or blue eyes.-2 These traits are merely speculated as emerging from a mutation long ago but persist in the present as trait options called gene variants or alleles.-3. Claiming such hereditary traits as being derived by a distant mutation assumes that Universal Common Descent is factual, and therefore these changes “must” have happened by mutations. These are merely speculations on the evidence and not what is empirically known regarding the evidence.
Summary: The gene sequences that eye coloration are known to be genetically passed as variants derived from the parents at conception (forming the various traits by gametes). As we have pointed out repeatedly, identifying any specific genetic sequence does not provide evidence for an ancient mutation–it just identifies the variants as they exist in the present. Heredity emerges from the sex cells of the parents that provide variants the ultimately form gametes that create the offspring at conception. This process has nothing to do with copy error mutations in the present. The assumption that eye pigmentation is due to mutation is based on a supposed ancient mutation (that was never observed by assumed). This claim is not empirical because no complete DNA sample exists from 6,000-10,000 years ago (of course). Any assumption of this occurring due to a beneficial mutation is based on speculation because, as far as we know, humans have always had various genes that affect eye coloration. Brown eye traits are dominant and blue eyes (or colorations that are not brown) are recessive. These gene variants have nothing to do with a copy error mutation in the present–only based on speculation.
1-https://www.sciencedaily.com/releases/2008/01/080130170343.htm;
2-https://www.nationalgeographic.org/encyclopedia/genotypes/;
3- https://www.yourgenome.org/facts/what-is-inheritance
“Lactose Tolerance Mutation”
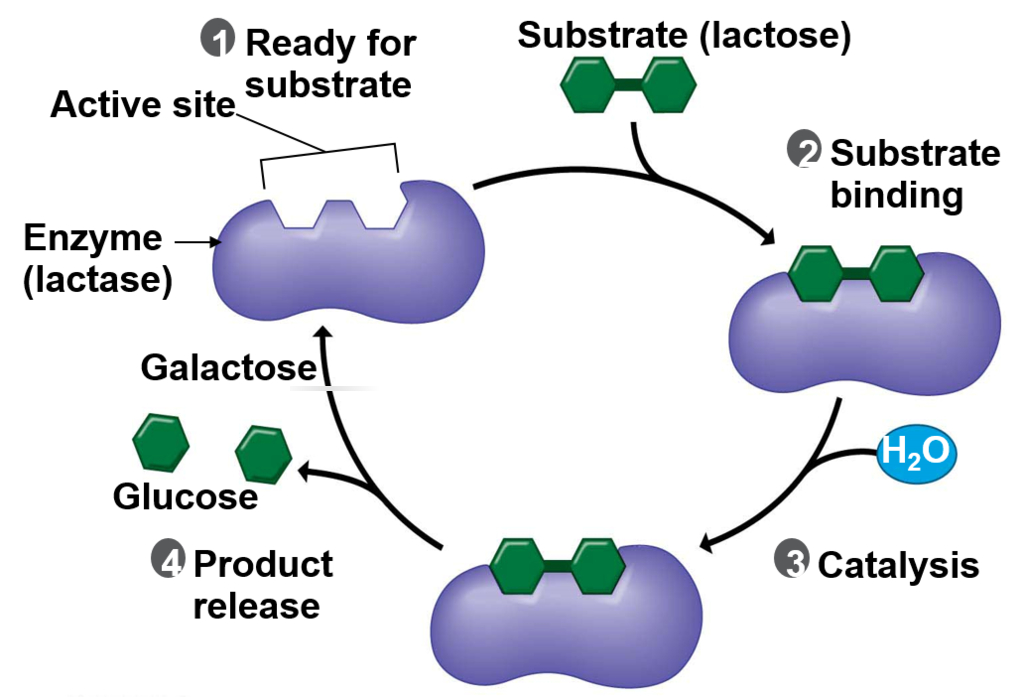
The Good: It is not hard to understand how digesting dairy (lactose) is a benefit. Evolution presumes that humans long ago developed the ability to digest dairy products by a mutation between 2,000 – 20,000 years ago from a region near modern-day Turkey. The idea concludes that humans domesticated cows, goats, and sheep.-1. Repeated exposure to the lactase enzyme ultimately caused a genetic mutation that enabled humans to digest dairy.
The reality: First, the biological mechanism that enables lactose digestion is by a (very complex) feedback loop—not by any single mutation (see diagram). Second, lactose is a sugar broken down by an enzyme called lactase provided by heredity (lactose intolerance is a recessive trait-2). Third, all humans become more susceptible to lactose intolerance after being weaned.-3 Forth, while fragments of DNA have been discovered, any genetic claims from thousands of years ago rely only on assumptions because isolating any fully mappable samples of DNA, both before and after the mutation, simply do not exist empirically. In other words, fifth, this proclaimed beneficial mutation is only speculation and not supported by any verifiable evidence. Finally, the reality seems to be more correct than lactose intolerance is the mutational disability.
Summary: The biological mechanisms behind lactose digestion are complex using a feedback loop that breaks the molecular bond by a highly specialized enzyme called lactase. This enzyme separates the sugar from the lactose molecule so it can be digested. Humans are born to digest lactose because they are fed breast milk from their mothers. As we age, the genetic expressions behind lactose digestion fade. The rate of this recession varies between individuals, but most all humans are less capable of lactose digestion than they were at birth. The idea that lactose tolerance emerged by a mutation fails superficial scrutiny. If human beings could not digest lactose from the beginning (from breast milk), then the species would have perished. The biological system has nothing to do with any mutation but a feedback loop. This supposed beneficial mutation of lactose tolerance developed in antiquity is falsified. The assumption that lactose tolerance developed from a prior intolerance is nonsensical. Like eye coloration in humans, this claim is not empirically established due to a mutation because no complete DNA sample existed from 6,000-10,000 years ago (of course). Any assumption of this occurring due to a beneficial mutation is based on speculation because, as far as we know, humans always could digest lactose. If not, we would not be here!
3-https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4586575/;
“Super-human Vision Mutation” (TETRACHROMATIC VISION)

The Good: This mutation claims those affected with the genetic mutation gain a “super-human vision” called Tetrachromatic vision. TetraChromatic vision (meaning the capacity to view four wavelengths instead of only three) means. At the same time, the average human can distinguish about one million colors. Those that carry the mutation claim might be capable of seeing 100 times more color variations. A stu y in The 2010 Journal of Vision Study found up to 12% of women had the genetic mutation is passed down from a parent that also carried the gene for color blindness.
The reality: Emerges from red-green color blindness in parents are passed usually to female offspring.-1 The 2010 Journal of Vision Study found exactly one woman (who claimed to have) tetrachromatic vision. It is unverifiable to confirm the claims of the color variations as there are no lab instruments to verify the claim empirically. Today, there are no established empirical methods to verify whether an individual has a “tetrachromat genotype” or not, let alone what additional colors they might be able to distinguish.-2 The researchers pointed out the obvious: “The evidence for strong tetrachromacy—where four independent cone signals are available for cortical transformations—is still elusive.” Neuroscientist Dr. Gabriele Jordan identified a single woman (subject cDa29) who could detect a greater variety of colors than trichromats could, corresponding with a functional tetrachromat (or true tetrachromat).-3
Summary: Out of 24 individuals used in the “largest” study, confirmed to be carrying this bonafide lab-verified mutation, only one woman claimed to have this super-human vision capacity. Interestingly, the mutation is usually passed to women from parents afflicted with color blindness. Seeing there are currently no means to empirically and objectively measure these claims, this is merely anecdotal. The researchers pointed out the obvious: “The evidence for strong tetrachromacy—where four independent cone signals are available for cortical transformations—is still elusive.” This supposed beneficial mutation is closer to mythology than any verified scientific genetic characteristic.
1-https://bigthink.com/surprising-science/evolution-is-still-happening-beneficial-mutations-in-humans/
2-https://jov.arvojournals.org/article.aspx?articleid=2191517; 2010 Journal of Vision Study
3- https://en.wikipedia.org/wiki/Tetrachromacy
“The Lenski E. Coli citrate digestion Mutation”

The Good: As an attempt to demonstrate evolution, since 1988, Richard Lenski has tracked mutations in E. coli bacteria by 2017. They had followed over 73 thousand generations and 10 thousand transfers. E. coli is typically unable to grow on citrate when oxygen is present. In 19 8, a mutated bacteria generation “gained” the ability to grow on citrate when oxygen was absent! This was “evolution” in action! An en entirely new fitness attribute gained (possibly) due to a mutation. Each culture used a little glucose but lots of citrates. Lenski hoped to force evolution to happen—force the bacteria to grow on citrate. In the 31,500th generation, a mutation seemed to provide evolution as a beneficial fitness gain in growth in citrate metabolism.-1
The reality: The New Scientist pointed out that E. coli never developed a new ability to metabolize citrate. It always could do this. It turns out that the citric acid, tricarboxylic acid (TCA), or Krebs regulates citrate’s expression (metabolism) as an energy source under anaerobic conditions, just not aerobic. The mutation jammed this operon’s regulation (expression) as a fault. The mutation likely caused a loss of specificity that equaled a loss of information. Mutations are good at destroying things, not creating them.2
Summary: While the study initially gained great notoriety, the resulting generations behind the mutation did not gain any new function other than broken switches. It turns out that bacteria have biological machinery that permits the consumption of sugars or citrates. Today it is well understood that the mutation was not evidenced for common descent evolution, nor was it evidence for a fitness gain. It was evident that the mutation that controls the switching between sugar and citrate digest broke.3 Lenski’s mutated bacteria, from a preexisting bacterial strain was certainly no novel organism, nor a new species as originally claimed. It has been established that this mutation causes a broken switch expression3 in the preexisting (and once fully functional) gene that only subjectively renders this as “beneficial”. Ultimately, these mutated bacteria can never revert to an oxygen environment and resume consuming glucose like normal bacteria. Why? Because their expression switch has been BROKEN by this mutation. Broken genetics are poor examples to defend common descent evolution. While counterintuitive, sometimes broken things provide benefit, they are still broken in the end. No one in their right mind can argue the transmutation from bacteria to humankind by failing genetics–it is preposterous.
…the implications of the work are often blown seriously out of proportion by a cheerleading science news media eager for stories to trumpet…the ability was due to the duplication and rearrangement of a gene for a protein that normally imports citrate into the cell, but only when no oxygen is present. The mutation allowed the protein to work when oxygen was present. (The switch was broken- no new “evolution”). Paranthesis, italics, and bold are mine.-4
Richard Lenski and Citrate Hype — Now Deflated, Michael Behe, May 2016
1-https://en.wikipedia.org/wiki/E._coli_long-term_evolution_experiment
2-https://creation.com/bacteria-evolving-in-the-lab-lenski-citrate-digesting-e-coli
3- “Citrate Death Spiral” Behe, Michael JUNE, 2020 https://www.discovery.org/a/citrate-death-spiral/
4- https://evolutionnews.org/2016/05/richard_lenski/
“Anti-Biotic Resistance”

The Good: Antibiotic resistance is proclaimed as an observable consequence of evolution (via natural selection). The antibiotic action is an environmental pressure; those bacteria which have a mutation allowing them to survive will live on to reproduce. They will then pass this trait to their offspring, a fully resistant generation.-1
The reality: Causes for antibiotic resistance in bacteria can be easily understood by beginning with what causes resistance. Bacteria are invaders, and antibiotics are developed to instruct our built-in immune system to kill these invaders. The antibiotic action is an “environmental pressure” because everything is working to eradicate the bacteria. Only those “lucky” few that “gained” a genetic mutation often render them invisible to the hard-working t-cell hitmen. This nadvertently permits survival and replication of the mutated.-2 Thereby, these mutated bacteria pass this mutated sequence to their offspring, which can emerge as an antibiotic-resistant generation.-2 The marvel here is not the copy error mutation, but the immune system and its incredible ability to kill invaders by specific genetic code translated by the t-cells.-3 Antibiotic resistance is a great example of how mutations invaders escape certain death by luck of a random genetic mutation but not strong evidence for universal common descent evolution.
Summary: If antibiotic resistance is adequately understood, it provides an abysmal example supporting universal common descent evolution. Bacteria can be good or bad within the bodies of living organisms. When harmful bacteria enter the body, a myriad of biological weaponry is deployed by our immune system to identify (by genetic sequence), find, and destroy these invaders. Our immunity, not the mutated harmful bacteria, is the true marvel. Some bacterial attacks have been remedied by artificial vaccines designed to help our immune systems eliminate these invaders. The vaccines (like our natural immune systems) receive the “hit” orders by a specific genetic sequence. This information is used by many biological functions, including T-Cells that find and destroy these specific genetically sequenced harmful bacteria. In the end, only the very few bacteria that had been mutated, changing the genetic sequence, are invisible to the “hit.” Now, these survivors multiply and become prevalent. Logically, it is no surprise that the original vaccine, like our own immune system, is often rendered ineffective against this new genetically varied strain–but not for long. The process merely repeats itself over and over–just as it has since the beginning of time. It is factually accurate that mutations occur in nature, and it is also true that they happen in bacteria. However, like all measurable mutations, they are due to entropy or broken genetics. Here, as with many other examples of broken genetics, it can benefit. However, considering antibiotic resistance as a prime example for universal common descent evolution or beneficial gains is ill-founded and based on a misunderstanding of biology and the immune system.
1-https://www.sciencedaily.com/terms/antibiotic_resistance.htm;
2-https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2937522/;
3-https://www.ncbi.nlm.nih.gov/books/NBK10762/;
4-https://biologywise.com/beneficial-mutation
The HIV Immunity mutation
THE GOOD: Thought to be a mutation from about 2,500 years ago, it is found in 1 in 10 people today and stops the spread of HIV in carriers. The CCR5 gene consists of 352 amino acids. In this deletion, one amino acid is mutated. This mutation inhibits normal cellular function, including inhibiting the HIV virus and potentially other proteins from entering certain cellular surfaces. “…the presence of the mutant delta32 protein in the endoplasmic reticulum inhibits transport of the wild-type CCR5 protein to the cell surface via a trans-dominant mechanism. (This is a deletion mutation.) most strains of HIV use CCR5 to enter host cells; the deletion of both copies of the CCR5gene (not one copy) protects against HIV infection….Individuals who are naturally homozygous for the delta32 mutation, which abolishes CCR5 expression, are generally healthy and at no apparent disadvantage.”1
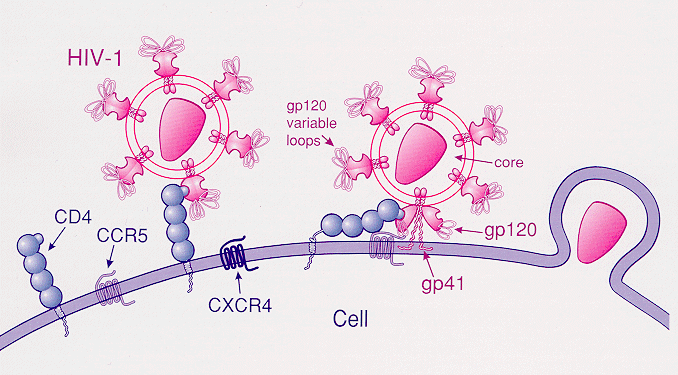
“However, apart from the protective effects against HIV infection, the impacts of this mutation, positive or negative, on other diseases are open to debate.”
Xie Y, Zhan S, Ge W, Tang P. The potential risks of C-C chemokine receptor 5-edited babies in bone development.-1
The reality: Linked to Alzheimer’s disease: “Although the mechanisms of Alzheimer’s disease are diverse and unclear, the past 20 years have witnessed the unprecedented development of the AD inflammation theory. As a key inflammatory receptor family, the C-C chemokine receptor family is a remarkable participant in the cause of Alzheimer’s disease; of this family, CCR5 is the most widely studied. CCR5 is essential for viruses to enter cells. The mutations inhibit HIV from entering the cells. It is thought this mutation might also impact other inflammatory and immune activities, but this remains to be largely unknown. New evidence on the inevitably intertwined link between Alzheimer’s disease and CCR5 indicates that CCR5 accelerates the development of Alzheimer’s disease, and few studies disputed it. The role of CCR5 in Alzheimer’s disease remains elusive. However, this intricate relationship will gradually be uncovered as the research progresses. Entanglement of CCR5 and Alzheimer’s Disease. Outside HIV, any additional “good” is unknown and causes memory deficits in mice: “Although the role of CCR5 in immunity and in HIV infection has been studied widely, its role in neuronal plasticity, learning and memory is not understood. Here, we report that decreasing the function of CCR5 increases MAPK/CREB signaling, long-term potentiation (LTP), and hippocampus-dependent memory in mice, while neuronal CCR5 overexpression causes memory deficits. CCR5 is a suppressor for cortical plasticity and hippocampal learning and memory, so its long-term effects might be detrimental.
Summary: The CCR5 Delta mutation is caused by the deletion of a single amino acid of the 352 within its protein chain construction. This mutation impacts yet largely (outside HIV research) unknown mechanisms of the function of T-Cells which fight severe disease. This variance works to prohibit HIV (and perhaps other diseases) from entering cells. This mutation has undisputed and inevitable links to Alzheimer’s Disease2although the exact role the mutation plays remains unknown. Seeing the normal function of this gene (without a mutation) is involved in highly critical functions of the cell regarding immunities its potential negative impacts when mutated remain unknown. This mutation is also linked to learning and memory disabilities due to CCR5 being a suppressor for cortical plasticity.3 Logically, this mutation might also prove to be dysfunctional as it also impacts normal biological functions as well. Outside benefits in stopping inflammatory responses and HIV infection, any “good” are unknown. Supposing the mutation to have occurred 2,500 years ago is clearly deep-time speculation supported (outside known HIV benefits) to be only anecdotal. The “benefit” in protecting against HIV is clear, but it still remains to be subjective (only benefiting those stricken with the disease), and its attributes outside HIV may very well prove to be objectively degradative.
1- Xie Y, Zhan S, Ge W, Tang P. The potential risks of C-C chemokine receptor 5-edited babies in bone development. Bone Res. 2019 Jan 29;7:4. doi: 10.1038/s41413-019-0044-0. PMID: 30701110; PMCID: PMC6351561.
2- Entanglement of CCR5 and Alzheimer’s Disease; Aging Neurosci., 07 August 2019
Sec. Alzheimer’s Disease and Related Dementias; Li, Tianwen Li & Zhu, Jianhong Aug 2019; https://doi.org/10.3389/fnagi.2019.00209
3- Zhou M, Greenhill S, Huang S, Silva TK, Sano Y, Wu S, Cai Y, Nagaoka Y, Sehgal M, Cai DJ, Lee YS, Fox K, Silva AJ. CCR5 is a suppressor for cortical plasticity and hippocampal learning and memory. Elife. 2016 Dec 20;5:e20985. doi: 10.7554/eLife.20985. PMID: 27996938; PMCID: PMC5213777.
“Viruses display evolution by mutation”
The Good: This is perhaps the number one claim made by Naturalists (including evolution popularizers) that proclaim mutating viruses are excellent examples for “evolution” in action. They claim the mutation process in viruses, forming them into different variants (altered genetic sequences by mutations), demonstrating Universal Common Descent evolution (UCD) right before our eyes as variants emerge. This claim is widespread.
The reality: Causes for antibiotic resistance in bacteria can be easily understood by beginning with what causes resistance. Bacteria are invaders, and antibiotics are developed to instruct our built-in immune system to kill these invaders. The antibiotic action is an “environmental pressure” because everything is working to eradicate the bacteria. Therefore, only those “lucky” few that “gained” a genetic mutation often render them invisible to the hard-working t-cell hitmen. This inadvertently permits survival and replication of the mutated.-2 Thereby, these mutated bacteria pass this mutated sequence to their offspring, which can emerge as antibiotic-resistant. The marvel here is not the copy error mutation but the immune system and its incredible ability to kill invaders by specific genetic code translated by the t-cells.-3 Antibiotic resistance is an excellent example of how mutations invaders escape certain death by happenstance of a random genetic mutation-4 but not strong evidence for universal common descent evolution.
Summary: The concept of universal common descent evolution (UCD) is (obviously) based on life. The idea is that “simple” living organisms such as bacteria are transmitted to humans over millions of years. The first problem with viruses being good evidence for UCD is viruses are not alive. They are likely packets of genetic information that escaped once fully functions cellular life. Today they are likely just parasitic genetic debris. The second problem is the chicken or the egg paradox of how viruses would have first emerged. While many imagine that perhaps non-living viruses provided a bridge to the first living organism, such conclusions are paradoxically creating a clear chicken or egg dilemma. Because viruses can only metabolize and replicate inside a live host, that host must have predated the virus itself. Like everything that contains genetics, mutations do occur. However, just as we have found in every conceivable case, modifications are destructive. Mutations cause entropy to operate as a form of unraveling. This is precisely what we observe in viruses. Viruses are bad examples for UCD but are excellent examples of devolution derived from broken genetics. This mutation can be claimed to be a fitness benefit similar to The Sickle Cell mutation. How so? While these conditions did and do provide survival fitness for those affected by deadly diseases, any benefit outside these fortuitous do not objectively exist. Occasionally broken things, counter-intuitively, can provide a subjective benefit. However, claiming such broken genetic functions as driven by mutations are strong evidence for universal common descent evolution is perposterous.
1- https://cosmosmagazine.com/science/biology/what-came-first-cells-or-viruses/;
3-https://www.ncbi.nlm.nih.gov/books/NBK10762/;
4-https://biologywise.com/beneficial-mutation
“Nylon consuming bacteria”

The Good: “Nylonase” is an example of a beneficial mutation in bacteria of the Flavobacterium strains. The nylonase bacteria was found in wastewater around manufacturing plants eating nylon molecules (nylon-6). The mutation in these bacteria involves the insertion of a single nucleotide in the genetic material. It is estimated that this frame-shift mutation first identified in 1975 was thought to have emerged in the 1940s when nylon was invented. Nylonase can be used in wastewater treatment plants.-1 The bacteria had a “nylonase” effectively digests chemicals which (was claimed) to have arisen by a random insertion “frame-shift” mutation, which formed a “stop” codon early in the gene, which caused a new “start” codon. The n w reading frame ran for 392 amino acids before the first “stop” codon, producing a large, novel protein!-2 This was the best example for Universal Common Descent Evolution yet!
The reality: Where did the nylon-eating ability come from? Carbohylesterases (been around much longer than the 1940s) are enzymes with broad substrate specificities meaning they can carry out a variety of reactions. Their binding pocket is large and accommodates many different substrates, including those hydrolyzed by nylonase. Tests revealed that various strains of Flavobacterium likely could hydrolyze such substrates well before the invention of nylon. Nylon is a by-product of petroleum that has been present for eons. The mutation here is called “frame-shift,” also called a “nonsense mutation” because it is improbable to result in anything useful. Indeed, the origin of nylonase provides an excellent example of the optimization of a preexisting protein fold but NOT the innovation or creation of a novel (all new) fold. -2
Summary: While the idea of a manufactured substance being consumed by a new bacterial organism seems convincing enough, it does not withstand scrutiny. First, nylon is made from petroleum, a natural and organic compound. Second, the identified mutation in the genetic sequence is due to a “nonsense” mutation that essentially never results in anything useful. Third, the dates of the emergence of this fitness gain are highly speculative and match the storyline but not necessarily the evidence (which is lacking). Finally, the original assumption that this was a new digestive gain of function has been disproven. Today, it has been well established that “nylonase” (bacteria capable of consuming nylon) did not “emerge” due to any sort of new gain of function mutation but was derived from preexisting genes within preexisting protein folds. Like the citrate-consuming bacteria, these bacteria already carry the genes to consume petroleum-based products, including nylon. There is nothing here that supports universal common descent evolution. This is a function of genetics passed by reproduction–this has nothing to do with copy error mutations.
1-https://biologywise.com/beneficial-mutation;
2- https://evolutionnews.org/2017/05/the-nylonase-story-when-imagination-and-facts-collide/
“Almond Tree non-bitter taste mutation”

The Good: Almond seeds from wild species contain amygdalin, a bitter chemical that converts into cyanide inside the human body. According to researchers, consuming wild almonds is fatal. A single gene mutation in wild almond trees resulted in a variety that no longer synthesizes amygdalin. When umans discovered this non-bitter almond species, they cultivated them, which is continued till today.-1
The reality: The emergence of this variant is due to genetic expressions at reproduction and not any mutation. The wild strain already carried the gene without amygdalin that was passed to the offspring forms used today for commercial almond cultivations. This is an example of a miscategorized “mutation” associated with gene variants provided by gametes are reproduction that is preexisting and not de novo (novel) variations due to mutations.
Summary: The genes for the bitterness variety of almonds preexisted from the sex cells of the parents (alleles). This has nothing to do with mutation. Geneticists have identified the sequence associated with this variety, but this does not equate to an empirical mutation. As with so many supposed ancient mutations, it is presumed that this happened long ago. Again, there are not fully sequenced DNA molecules to confirm that this mutation occurred– this is speculated. What we know for sure is the variety of almonds comes from other preexisting gene variants of, you guessed it, other almond trees.
1-https://biologywise.com/beneficial-mutation
“Murray Gray Beef Cattle mutation”

The Good: Murray Gray is a cattle breed obtained accidentally from a traditional cow species. The c lves produced by the specific cow were more productive than those made by the others. Farme s soon noticed the difference and started breeding from the offspring. This ay, the Murray breed with some of the most favorable characteristics has become popular all over Australia, which then spread to various other countries.-1
The reality: The emergence of this cow species was rendered by natural selection due to genetic expressions at reproduction– not any specific mutation. The offspring with the most desired traits were isolated for commercial farming. This is an example of a miscategorized “mutation” associated with gene variants provided by gametes at reproduction. Gamet s are preexisting within the parents and were not created de novo (novel or new information) variations due to mutations.
Summary: The genes for the variety of cattle that ultimately formed that “Murray Gray” came from preexisted from the sex cells of the parents (alleles) formed at conception–not due to mutation. Like all the various breeds of dogs that emerged from the Grey Wolf, all the breeds of cattle come from other cows. Geneticists have identified the sequence associated with genetic variation, but this identification does not equate to an established mutation. As with so many supposed ancient mutations, it is presumed that this happened long ago. Again, there are not fully sequenced DNA molecules to confirm that this mutation occurred– this is speculated. Speculated based on the presumed starting point that universal common descent is correct. So, UCD proves an ancient UCD? This is a logical fallacy. We know for sure that the wide variety of almonds (like all other living organisms) comes directly from heredity. Gene variants are preexisting from the parents (the mommy and daddy cows). The Murray Gray Cattle is an isolated (bottlenecked) artificially selected breed derived by heredity, not mutation.
1-https://biologywise.com/beneficial-mutation
Mutational types referenced:

(a) VIRUSES. Virus s are proposed as good evidence for beneficial mutation because they mutate and modify. However, viruses are non-living lipid capsules of degraded genetic material. Virus s also are parasites requiring a host for metabolism and replication. Most are harmless and are likely genetic debris that remains from a once fully functional genetic process;
(b) ANCIENT. Unverified mutations speculated to have first happened long ago, such as lactose tolerance. These are not empirically established but are supposed to have occurred compared to preexisting genetic material. Then, these alterations become the speculation behind presumed mutations of the past. The specific genetic region that affects these attributes is known in the present, but knowledge of the preexisting genetic sequences does nothing to prove a past ancient mutation. Pre-mutational data of a fully sequenced DNA molecule simply does not exist;
(c) ANECDOTAL These have grand claims but often very few examples to confirm if any. They usually are untestable or verifiable by modern lab equipment. Many attributes can be spun as beneficial or a fitness gain, but it is anecdotal when data is lacking. A few examples are tuskless African Elephants or tetrachromatic vision.
(d) HEREDITARY & EPIGENETIC EXPRESSIONS These are miscategorized because they are not due to mutations! These are characteristics associated with preexisting genetic gametes (dominate and recessive traits) related to heredity. These are proposed as beneficial mutations but result from preexisting reproduction functions. Some examples are fewer wisdom teeth or divers holding breath “mutations.”
(e) LAB VERIFIED. Final y, lab-verified mutations with living organisms with evidence from both (i) the original and (ii) mutated traits outside gametes. Examp es include “unbreakable” (stronger) bones, higher good cholesterol levels, increased oxygen capacity, fast-twitch muscle fiber that might provide the athlete to be quicker and more robust, and a handful of others. Note: merely identifying the gene responsible for the trait is not identifying the mutation because the data for the preexisting genetic sequence must be validated otherwise it is simply an assumption (usually “ancient”).